
python commands
Published:
useful python commands
large data by bytes
import numpy as np
import pandas as pd
from scipy.stats import pearsonr, spearmanr
n_processors = 4
gct = 'your_file.gct' # replace with the actual file path
def scan_linepos(path):
"""
Scan the file and store the byte offsets for each line.
"""
linepos = []
offset = 0
with open(path) as inf:
for line in inf:
linepos.append(offset)
offset += len(line)
return linepos
linepos = scan_linepos(gct) # the scan only needs to be done once
linepos = linepos[3:] # remove the header lines, adjust depending on the file format
def sample_lines(linepos, l0=None, path=gct):
"""
Sort the line positions for efficient file reads.
"""
linepos.sort() # this may make file reads more efficient
linepos = pd.DataFrame({0: linepos})
return linepos
def _corr(offset1, l0=None, path=gct):
"""
Calculate Pearson and Spearman correlation between two sets of values.
"""
with open(path) as inf:
inf.seek(offset1)
l1 = inf.readline().split("\n")[0] # Read the line
l = l0.split("\t") # Assuming l0 is a header or row for the comparison
l_ = l1.split("\t")
gid = l[0]
gid_ = l_[0]
l = l[2:] # Skip first two columns (Gene identifiers or metadata)
l_ = l_[2:]
tmp = pd.DataFrame({0: l, 1: l_}) # Create DataFrame to handle data
tmp = tmp[(tmp[0] != "") & (tmp[1] != "")] # Remove empty values
tmp = tmp.astype(float) # Ensure all values are float
tmp = tmp[(tmp[0] > 0) & (tmp[1] > 0)] # Filter out non-positive values
n = len(tmp)
if n > 2:
tmp = np.log10(tmp) # Log-transform the values for correlation
l = tmp[0].tolist()
l_ = tmp[1].tolist()
pearson_stat, pearson_p = pearsonr(l, l_)
spearman_corr, spearman_p = spearmanr(l, l_)
res = [gid, gid_, n, pearson_stat, pearson_p, spearman_corr, spearman_p]
else:
res = [gid, gid_, n, None, None, None, None]
res = [str(s) for s in res] # Convert to strings for output formatting
res = "\t".join(res) # Join all results with tab
return res
def process_lines(linepos, l0=None):
"""
Process the lines to calculate correlations for all pairs.
"""
linepos = pd.DataFrame({0: linepos}) # Convert to DataFrame for easy manipulation
results = []
for idx, row in linepos.iterrows():
offset1 = row[0] # Get the offset for the line
result = _corr(offset1, l0) # Get the result for this line
results.append(result)
return "\n".join(results) # Join all results with new lines for final output
# Run the processing function and print the results
results = process_lines(linepos)
print(results)
seaborn
from matplotlib.backends.backend_pdf import PdfPages
import matplotlib.pyplot as plt
import seaborn as sns
# Assuming pairs_set, all_betas, matrix_betas, project_folder, mle_matrix, and new_name are defined
pairs = [list(pair) for pair in pairs_set]
# label look like ['matrix(Time|beta)', 'pairwise(JEKO_Cyta|beta)'
def label_pair(pair):
first_element = f"matrix({pair[0]}|beta)"
second_element = f"pairwise({pair[1]}|beta)"
return [first_element, second_element]
label_pairs = [label_pair(pair) for pair in pairs]
#plot
font = {'family' : 'serif',
'weight' : 'normal',
'size' : 16}
pdf = PdfPages(project_folder+"/"+mle_matrix+"/pairwise_vesus_matrix_correlationPlots"+new_name+".pdf")
w=4
l=3
i=1
fig = plt.figure(figsize=(30,20))
for pair in label_pairs:
x=pair[0]
y=pair[1]
df_plot=all_betas[['Gene']+[y]].merge(matrix_betas[['Gene']+[x]], how="right", on="Gene")
ax = fig.add_subplot(l,w,i)
#ax.set_title("%s" %(gene_name), font)
sns.scatterplot(x=x, y=y, data=df_plot)
ax.set_xlabel(x, font)
ax.set_ylabel(y, font)
ax.tick_params(labelsize=16)
#matplotlib.rc('axes', linewidth=2)
i=i+1
if i == w*l+1:
plt.tight_layout()
plt.savefig(pdf, dpi=300, bbox_inches='tight', pad_inches=0.1,format='pdf')
plt.show()
plt.close()
i=1
fig = plt.figure(figsize=(30,20))
if i != 1:
plt.tight_layout()
plt.savefig(pdf, dpi=300, bbox_inches='tight', pad_inches=0.1,format='pdf')
plt.show()
plt.close()
pdf.close()
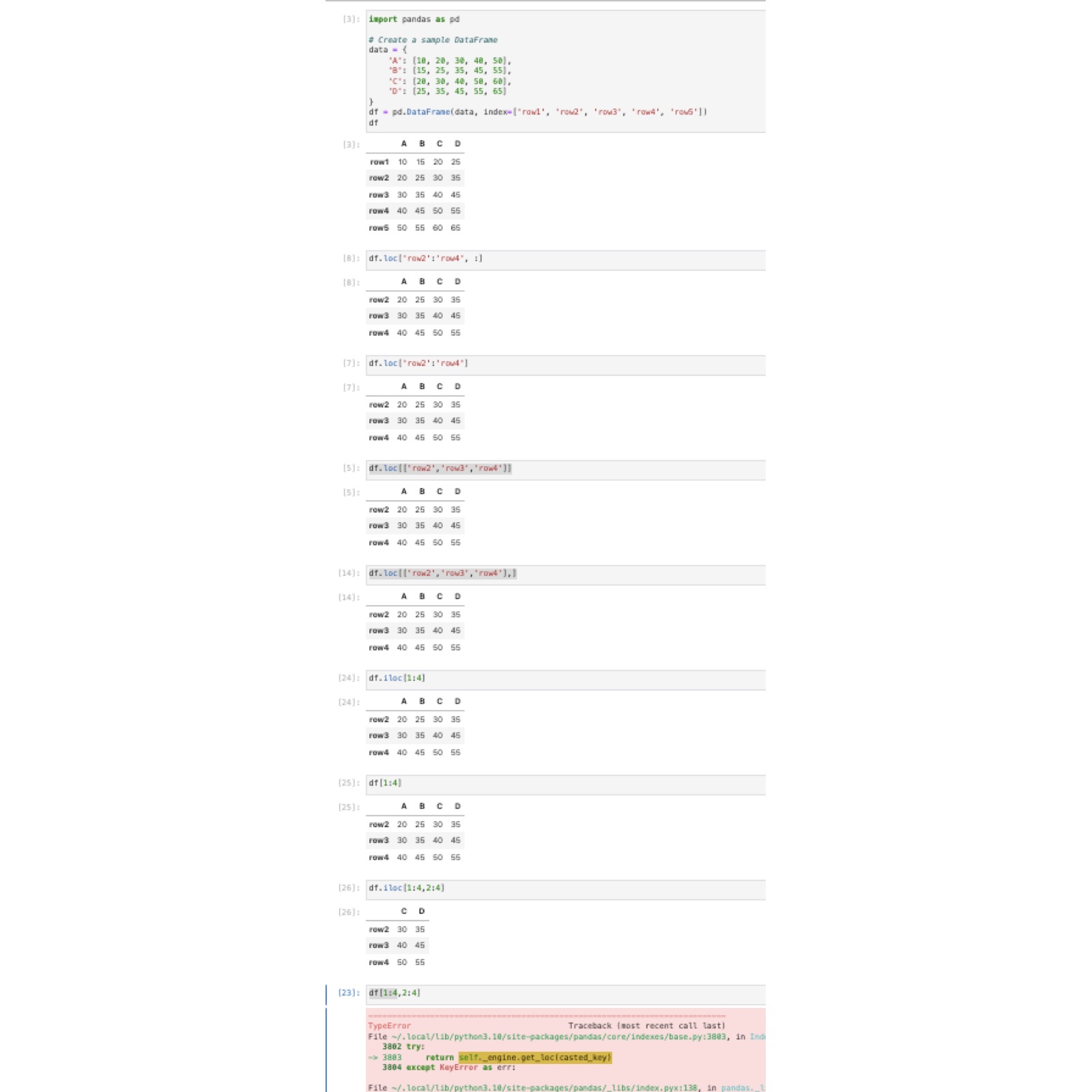
loc iloc
df.loc[["row2", "row3"], :]
df.loc[["row2", "row3"], ["A", "D"]]
df.iloc[1:4, :]
df.iloc[1:4, [0, 3]]
{kind=link}
display whole dataframe
from IPython.display import display, HTML
display(HTML(df.to_html()))
combine lines with same pattern/prefix
prefixes = set()
##Read data from file
with open('list.txt', 'r') as file:
lines = file.readlines()
##Collect unique prefixes
for line in lines:
prefix = line.strip().split('_')[1]
prefixes.add(prefix)
##Sort prefixes
sorted_prefixes = sorted(prefixes)
##Concatenate lines with the same prefix
output = []
for prefix in sorted_prefixes:
matches = [line.strip() for line in lines if line.strip().split('_')[1] == prefix]
combined = ', '.join(matches)
output.append(combined)
result = '\n'.join(output)
print(result)
input
a_123
b_456
a_789
b_101
c_112
output
a_123, a_789
b_456, b_101
c_112
jupter magic cell
%%bash
%%writefile